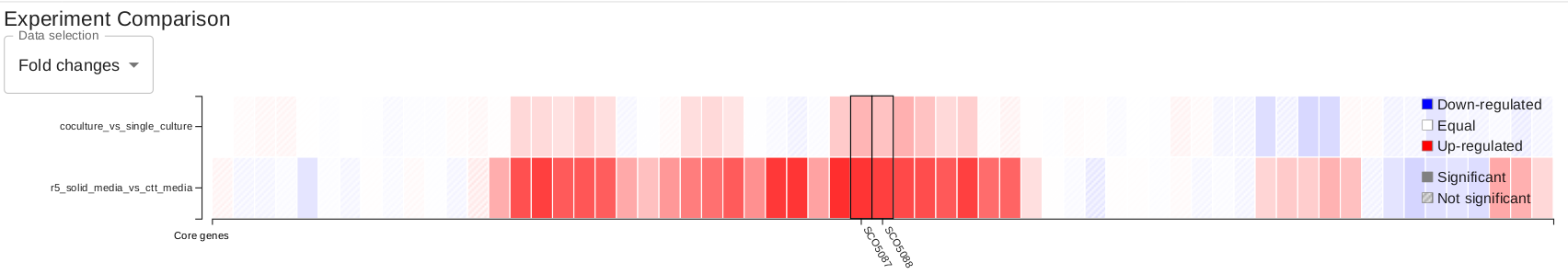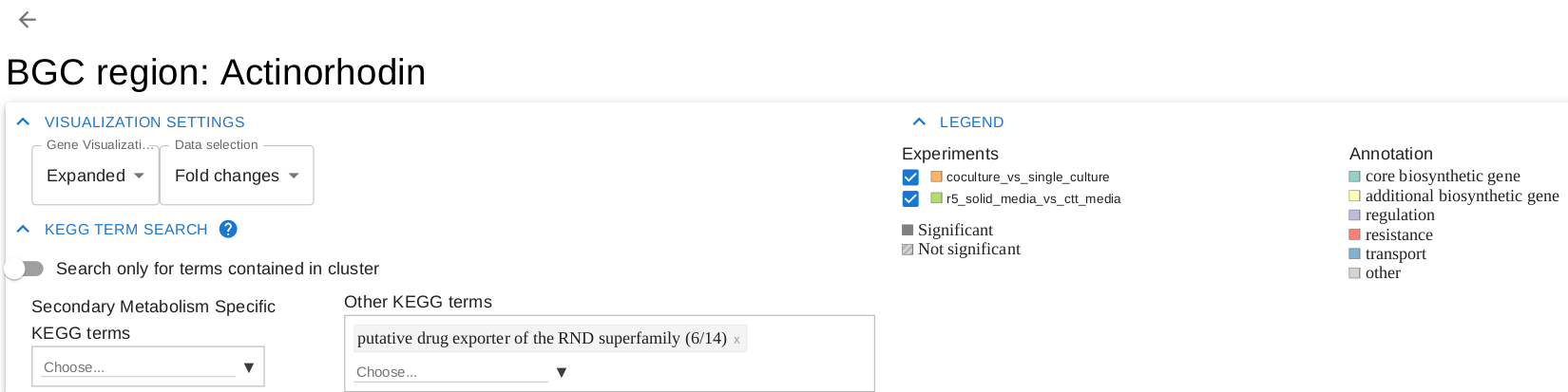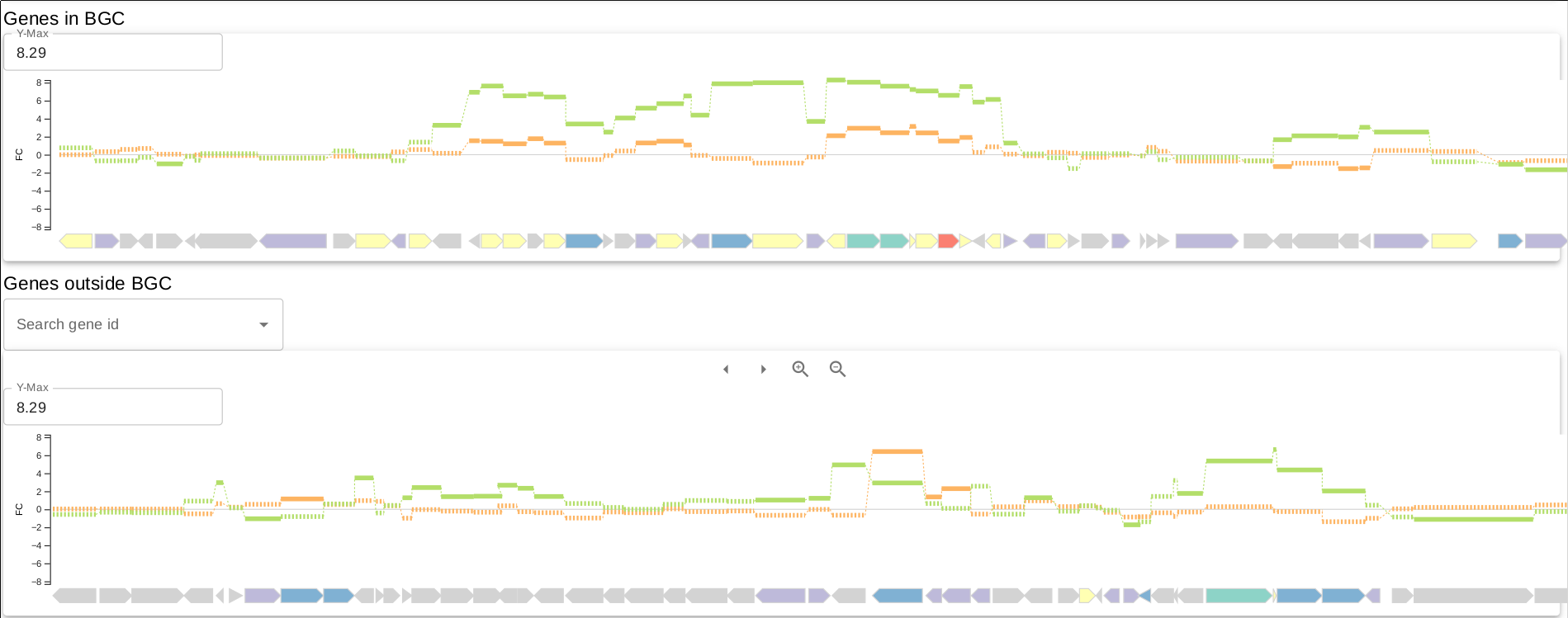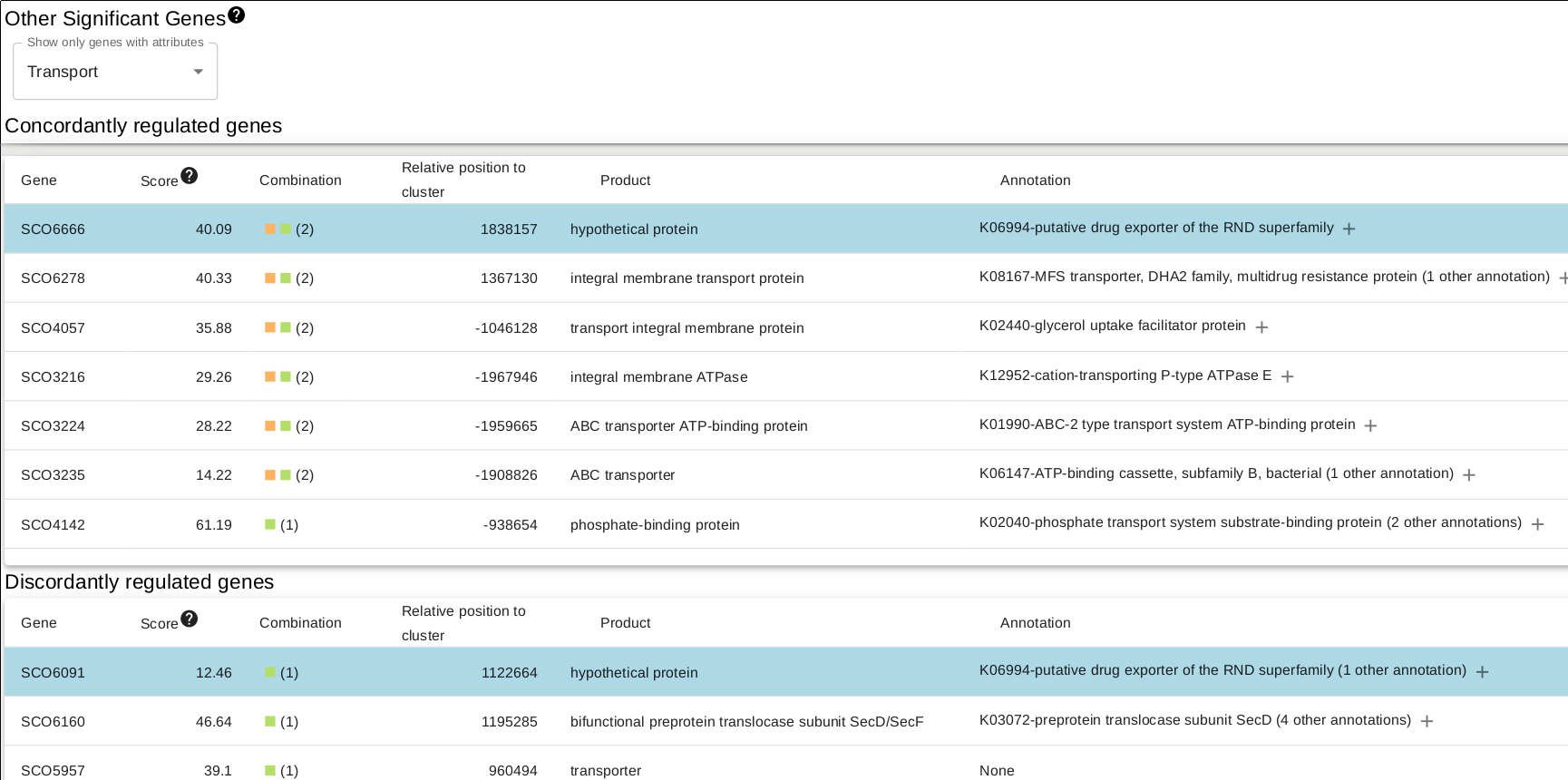
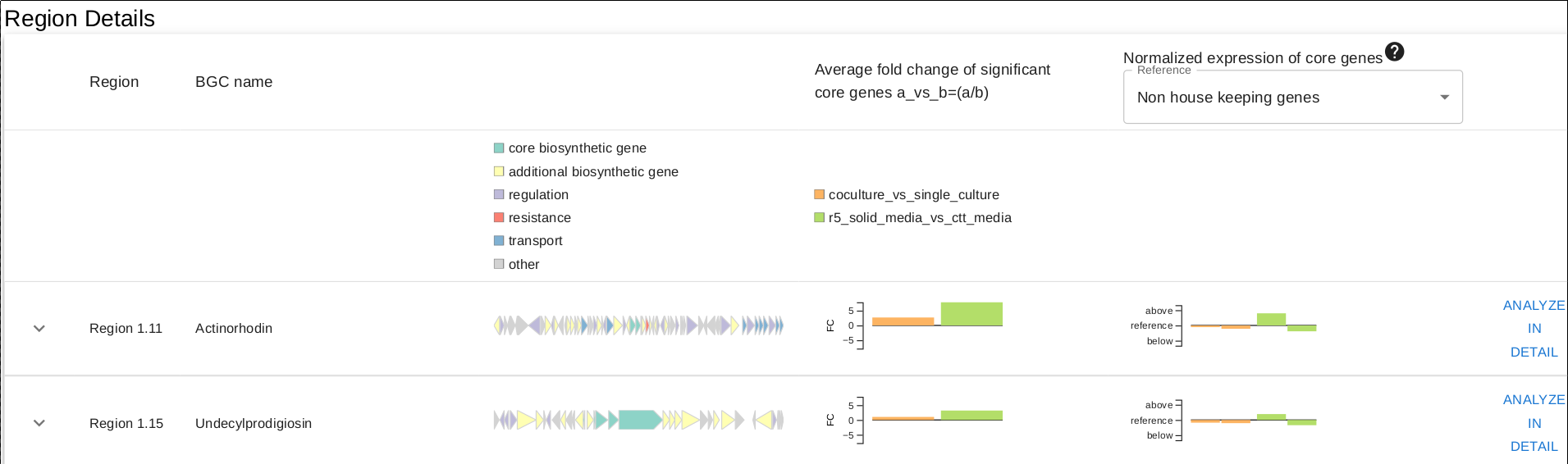
In the overview page, you will find expression information of detected BGCs based on your RNA-seq experiments. Overview can be sorted by the selected column title e.g. "Average fold change..."
- "Region" column denotes the region numbers given by antiSMASH.
- "BGC name" column denotes a similar (if similarity is detected by antiSMASH) BGC defined in MiBIG.
- Third column is the overview of genes residing in the BGC, pretty self-explanatory.
- Average fold change column simply shows a summary of the fold change (calculated from core biosynthetic genes) between conditions for each experiment for the respective BGC.
- Final column is a simple representation of the BGCs expression level, compared to specific references:
- "Housekeeping genes": Mean value of expression levels (TPMs) of all the housekeeping genes detected in the genome
- "Non housekeeping genes": Mean value of expression levels (TPMs) of all the non housekeeping genes detected in the genome
- "Mean expression": Mean value of expression levels (TPMs) of all the genes in the genome.